Sickle Cell Disease: Prevalence, Challenges And Road Map For Its Control In India By 2047
Prof. Hariom Sharma1

1Laboratory Director Govt. Medical College & Sir T. Hospital Bhavnagar – Gujarat – India Hon. Scientific Coordinator of SCDIO – France.
Corresponding Author: Prof. Hariom Sharma
E-mail: hariom_sharma2000@yahoo.com
APFCB News Volume 2, Issue 2
Different forms of Sickle Cell Disease (SCD) and its worldwide distribution
In Africa, Caribbean, North & South America, and Europe:
- Sickle Cell occurrence known as Haplotype
- Sickle hemoglobin (HbS)
- Sickle hemoglobin C (HbC)
- Beta thalassemia Sickle cell
- Homozygous Sickle cell (SS)
In Asia & Middle East:
- An additional Sickle Cell occurrence known as Asian Haplotype
- Shared by people in Asia, the eastern province of Saudi Arabia and throughout central India
- Asian Haplotype associated with high levels of fetal hemoglobin and frequent deletional alpha thalassaemia

United States
The exact number of people living with SCD in the U.S. is unknown, but it is estimated that approximately 100,000 Americans are affected by the disease. One out of every 365 Black or African-American is born with SCD. Whereas one out of every 16,300 Hispanic-American are born with the disease.It is reported that 1 in every 13 Black or African-American babies carries sickle cell trait (SCT)
Africa and Asia
Half of the world’s SCD population lives in three countries: Nigeria, India, and the Democratic Republic of Congo. The disease affects up to 2 percent of the population, and the carrier prevalence rate (sickle cell trait) is as high as 10 to 30 percent Nigeria alone has been estimated to have at least 150,000 newborns with SCD annually. Estimates are challenging because of the lack of federal newborn screening programs; however, approximately 700,000 births occur per year and the prevalence of SCD in newborns was 3 percent in a regional newborn screening program report.
India
India has the largest tribal population which is 8.6% /67 million of the total population. The prevalence of sickle cell carriers among different tribal groups varies from 1 to 40 per cent.
Distribution of Hb Variants in India
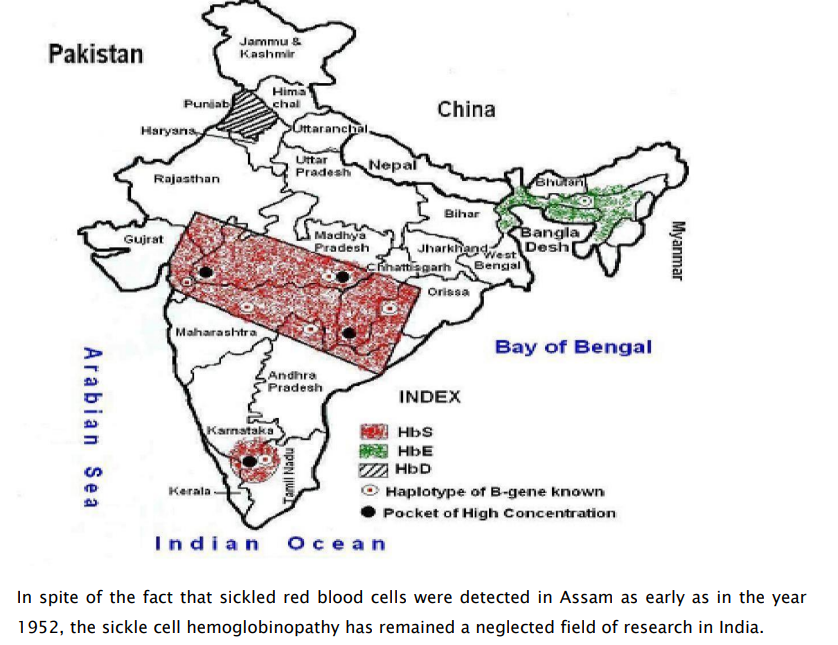
Challenges to control SCD
SCD is Illness of ignorance, poverty & pain, it is necessary to destroy the myths that are sources of indifference and passivity. The comparison of the therapeutic possibilities between developed and developing countries is an example of more than unbearable inequality; troubling evidence emphasizes the tragic disparity among humans facing disease and death. In the developing countries, financial burden on the parents of the children suffering from SCD limits the quality of the daily medical care and sometimes results in their death for lack of available but expensive cares such as transfusion or antibiotics prophylaxis.
Role of clinical laboratories and healthcare system in the management of SCD
Laboratories can play a vital role in screening of the population for the disease diagnosis, whereas Primary Healthcare/Tertiary care hospital shall have facilities for immunization, necessary medicine and infrastructure for daycare to be affordable. Transfusion facilities can help primarily in management of the SCD patients and also in bringing down the burden of the disease.
Prenatal diagnosis
Although there are significant advances in the management of sickle cell disease, yet increased morbidity and early death are not infrequent. Thus, prenatal diagnosis remains an important option for couples at risk of having a child with homozygous sickle cell anemia, sickle β thalassemia or HbSD disease despite the fact that it is impossible to predict the severity of the disease and many individuals may have a milder clinical presentation. With increasing awareness in the community more couples are opting for prenatal diagnosis. It is reported that around 400 prenatal diagnosis for sickle cell disorders about 20 per cent were tribal couples mainly from South Gujarat, Maharashtra, Madhya Pradesh and Odisha. Chorionic villus sampling at 10 to 12 weeks of gestation and DNA analysis by reverse dot blot hybridization or allele specific priming were the methods of choice for prenatal diagnosis. This makes prenatal diagnosis simpler and more cost-effective in tribal populations. For couples who came late, the cordocentesis and foetal blood analysis by HPLC were done. These technologies have now been established at regional centers.
Role of laboratory in monitoring and management of SCD patient
There are numerous methods available to diagnose hemoglobinopathies. The first is hemoglobin fractionation and consists of separating hemoglobin species based on charge and/or size. Hemoglobin fractionation can be performed by high-performance liquid chromatography (HPLC) methods, capillary electrophoresis (CZE) methods, and gel electrophoresis methods (alkaline and acid gels and isoelectric focusing, IEF gels). HPLC separates hemoglobin fractions based on charge, whereas with CZE and gel methods, separation is based on both size and charge. HPLC and CZE are high resolution quantitative techniques. Gel methods, on the other hand, are low resolution compared to HPLC and CZE, although they can be semi quantitative. Mass spectrometry and molecular methods (sequencing and deletion/duplication) are extremely powerful techniques for diagnosing hemoglobinopathies and thalassemia, particularly those that are challenging to diagnose with HPLC, CZE, and gel methods. However, these methods are for qualitative identification and do not quantify hemoglobin species. Patients with SCD have historically been managed by RBC transfusions and/or hydroxyurea. Managing and monitoring patients on these treatments requires discrimination and accurate quantitation of the various hemoglobin species; therefore, HPLC or CZE are the preferred methods for following:
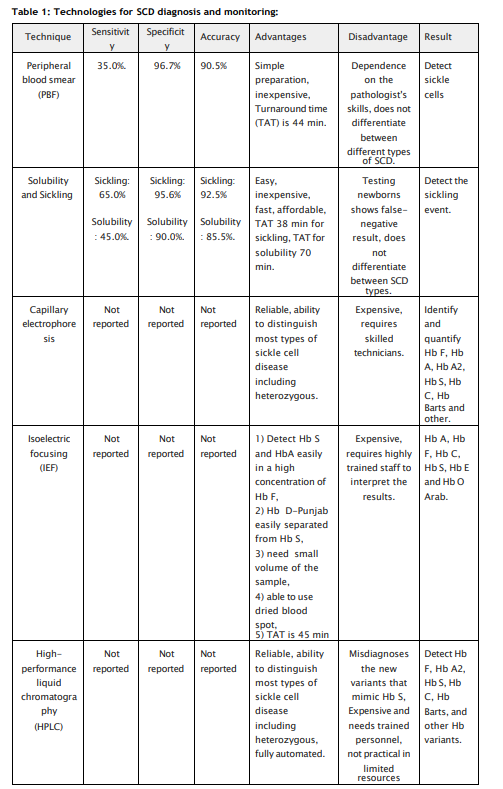
It is evident that there is availability of broad spectrum laboratory tools and techniques available for the mass screening of the population followed by confirmatory diagnosis and prognosis of the disease. A network of laboratories need to be created for the screening of population which is pre-disposed to SCD followed by strategic sickle cell control program planning and implementation will certainly will meet the nations goal to eradicate the disease
Road Map
The National Sickle Cell Anemia Elimination Program, introduced in the Union Budget 2023, focuses on addressing the significant health challenges posed by sickle cell disease, particularly among tribal populations of the country. Sickle cell disease (SCD) is a chronic single gene disorder causing a debilitating systemic syndrome characterized by chronic anemia, acute painful episodes, organ infarction and chronic organ damage and by a significant reduction in life expectancy. Implemented in 17 high-focus states
Across the country, this program aims to improve the care and prospects of all sickle cell disease patients while reducing the prevalence of the disease. The 17 states are- Gujarat, Maharashtra, Rajasthan, Madhya Pradesh, Jharkhand, Chhattisgarh, West Bengal, Odisha, Tamil Nadu, Telangana, Andhra Pradesh, Karnataka, Assam, Uttar Pradesh, Kerala, Bihar, and Uttarakhand. The program is executed in a mission mode as part of the National Health Mission (NHM), aims to eliminate sickle cell genetic transmission by the year 2047, showing a long-term commitment to eradicating the disease. Over a period of three years, spanning from the fiscal year 2023-24 to 2025-26, the program targets screening approximately 7.0 crore people. This ambitious goal highlights the program's dedication to reaching a large portion of the population, promoting early diagnosis and intervention. All these exaggerated efforts will certainly eradicate the sickle cell disease in the population.
1. Nicholas J Kassebaum, Institute for Health Metrics and Evaluation, Hans Rosling Center for Population Health, Seattle Lancet Haematol 2023; 10: e585–99:Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: a systematic analysis from the Global Burden of Disease Study 2021.
2. R. S. Balgir: Epidemiology, Population Health Genetics and Phenotypic Diversity of Sickle Cell Disease in India. The Internet Journal of Biological Anthropology. 2007; 1(2).
3. Roshan B. Colah, Malay B. Mukherjee, Snehal Martin, PMC Disclaimer Sickle cell disease in tribal populations in India. Indian J Med Research 2015; 141(5):509-15.
4. Okwi A.L., Byarugaba W., Parkes A.T.R. of S. and S.T. and P.; Ocaido, M. The Reliability of Sickling and Solubility Tests and Peripheral Blood Film Method for Sickle Cell Disease Screening at District Health Centers in Uganda A. Clin. Mother Child Heal. 2010; 7:1–5.
5. Frömmel C. Newborn Screening for Sickle Cell Disease and Other Hemoglobinopathies: A Short Review on Classical Laboratory Methods-Isoelectric Focusing, HPLC, and Capillary Electrophoresis. Int. J. neonatal Screen. 2018; 4: 39.
6. Nair S. Potential Pithfalls in Using HPLC and its Interpretation in Diagnosing HbS. J. Rare Dis. Res. Treat. 2018; 3: 9–12.
7. Wjdan A. Arishi, 1 Hani A. Alhadrami,1,2,* and Mohammed Zourob3,*Techniques for the Detection of Sickle Cell DiseaseMicromachines (Basel). 2021; 12(5): 519.
